Navigation
Neurodegeneration Products Dashboard
Aβ
tau
α-synuclein
TDP-43
C9ORF72
Huntingtin(this page)
Ordering Information
Neurodegeneration-related Flyers and Brochures
Antibodies for Neurodegeneration Research (TDP-43, 4R-tau, C9ORF, Synuclein)
PDF Download
Oxidative Stress Markers
mAbs and Selection Guide
PDF Download
Autophagy/Mitophagy Antibodies
PDF Download
Glutamate and NMDA related
Dopamine related
Cholesterol
Ganglioside
Glucosylceramide
Sphingosine
Mitochondria related
Heat shock
Oxidative stress
DNA damage
Lipid peroxidation
Protein carbonylation
Senescence
Transcription and Chromatin
Autophagy
Nucleocytoplasmic transport
Prion protein
APOETREM2
Progranulin
SOD1
FUS
Humanin
Gfap
Paraoxonase
CD36
Dysbindin
BDNF
VEGF
PARP
PPAR gamma
Ubiquitin
Proteasome
Proteases
Caspases
Peptidyl-prolyl cis-trans isomerases
NADPH oxidases
Dyneins
Kinesins
Kinases and Kinase inhibitors
Signaling molecules
Nuclear Hormone Receptors
Neurodegeneration Products: Huntingtin
| Catalog Number | Product | Reactivity | Unit size |
| CSB-EL010905HU | Human Huntingtin (HTT) ELISA kit | Human | 1x96 rxns |
Huntington disease (HD), first described in 1872, is an autosomal dominant, neurodegenerative disorder characterized by progressive movement, cognitive, behavioral and psychiatric abnormalities, with motor symptoms typically beginning between age 35-45 and death occurring 10-20 years after symptom onset. HD neuropathology originates with degeneration of striatal medium spiny neurons (MSNs) followed by white matter loss and widespread forebrain atrophy. Currently, there are no disease-modifying therapies for HD. In 1993 the genetic cause of HD was precisely located in the expansion of a polyQ-encoding CAG repeat tract in exon 1 of the chromosome 4 huntingtin (HTT) gene. The number of CAG repeats in HTT is polymorphic, occurring in the non-HD population between 9 to 35 times. A CAG expansion exceeding 35 repeats results in HD. Rare carriers of 36 to 39 CAG repeats have lower penetrance and later onset of the disease than those with 40 or more CAG repeats. Indeed, the age at onset of disease is inversely proportional to the length of the CAG expansion, with juvenile onset associated with HTT carrying about 75 or more repeats. HTT is ubiquitously expressed (at varying levels), found not only in neurons but also astrocytes and oligodendrocytes and many other cell types. Most HD patients carry one normal and one mutant allele. Because of its dominant inheritance pattern, HD was first thought to arise solely from toxic gain-of-function mechanisms. However, loss of HTT function also contributes to disease. Furthermore, mutant polyQ HTT (mHTT) exerts dominant-negative effects on HTT that impair its normal cellular functions in intracellular trafficking, transcription, autophagy, cell division, ciliogenesis, migration and lead to dysfunction of synaptic signaling, impaired protein folding and transport, alterations of gene transcription and translation, inhibition of protein clearance pathways, endoplasmic reticulum stress, mitochondrial disturbances, perturbation of Ca2+ signaling, amino acid metabolism deficits and cell death. Thus, it is likely that pathogenesis involves both gain and loss of function mechanisms. Please scroll to the ORDERING section to explore CosmoBio USA's Huntingtin reagents that have proven helpful in the study of neurodegenerative diseases.
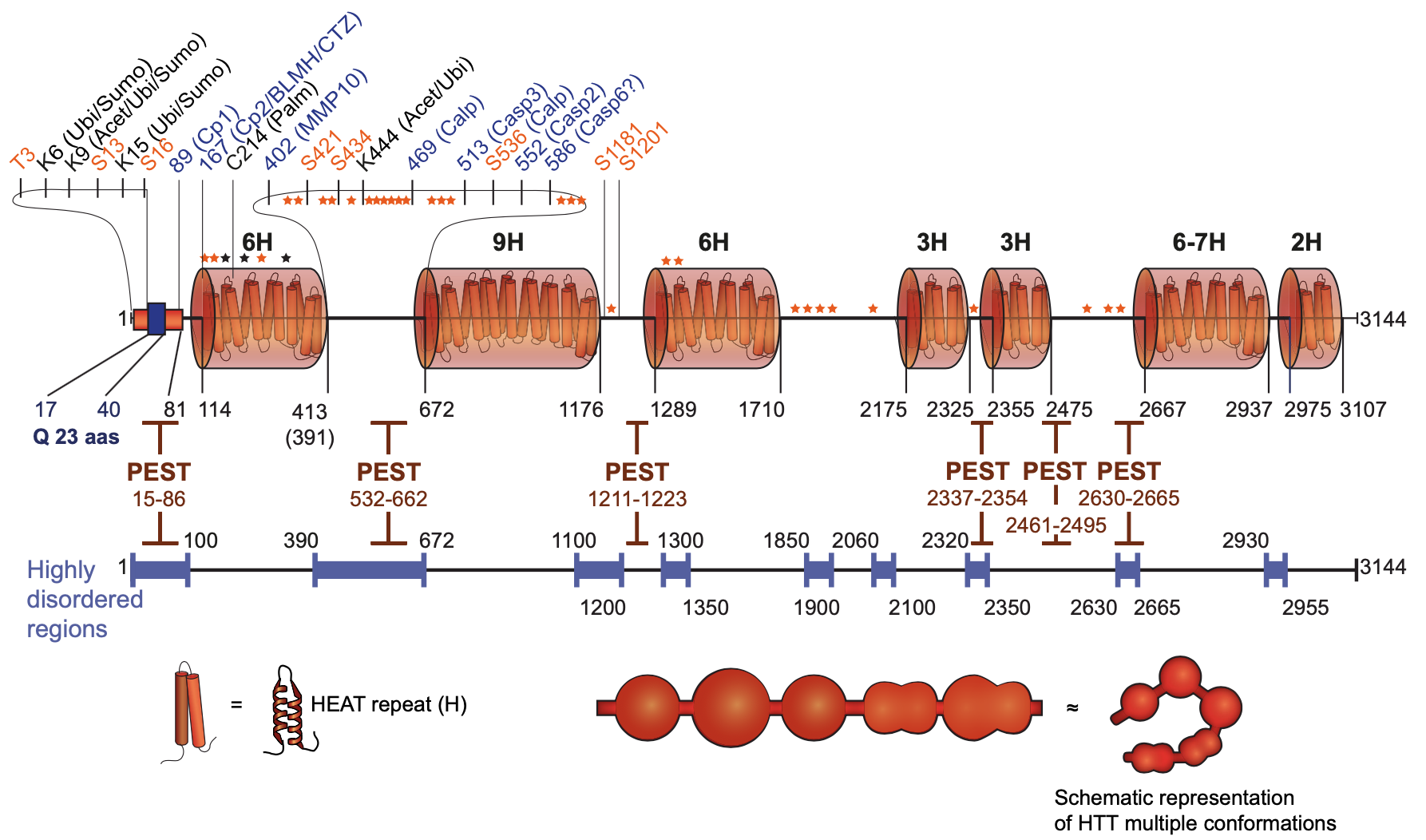 From: Saudou, F., Humbert, S. (2016). The Biology of Huntingtin Neuron 89(5), 910-926.
From: Saudou, F., Humbert, S. (2016). The Biology of Huntingtin Neuron 89(5), 910-926.
HTT is a 348-kDa, 3144 amino acid protein whose principal structural feature is a set of 7 clustered HEAT repeats, solenoid-like structures comprising two alpha helices linked by a short loop. The HEAT repeat domains likely function as scaffolds for binding protein complexes and to mediate inter- and intra-molecular interactions. The middle region of HTT (507–1,230) can bind to N-terminal (1–506) and C-terminal (2,721–3,144) domains; the 507–1,230 domain can also self-associate to form HTT homodimers. Similarly, N-terminal parts of HTT (1–416 and 1–586) bind to different C-terminal regions (1,725–2,800 and 2,416–3,144, respectively). These observations suggest that HTT can adopt various three-dimensional (3D) conformations, depending on its intra-molecular interactions. These interactions may also involve other protein complexes, as HTT has numerous interacting partners. In agreement, purified HTT can adopt up to 100 structurally distinguishable conformations. Supporting the functional relevance of HTT conformers, a conformation of mHTT recognized by monoclonal antibody 3B5H10 was degraded more slowly than mHTT conformers recognized by other monoclonal antibodies. Other functional motifs, such as a NES at position 2,397–2,406, further regulate HTT function and localization.
HTT undergoes proteolysis at multiple sites in dispersed PEST (standing for: proline, glutamic acid, serine and threonine) domains that occur mostly in highly disordered regions of the protein. These proteolytic sites are present in both HTT and mHTT; and, in vitro, mHTT is as good a cleavage substrate as HTT. HTT proteases include several caspases, calpain, cathepsins and the metalloproteinase MMP10. Importantly, in HD brains there is a specific increase in the activity of these proteases, leading to the generation of small N-terminal, polyQ-containing fragments that are translocated from the cytoplasm and toxically accumulate in the nucleus. In addition, C-terminal fragments, that do not contain the polyQ stretch, induced toxicity via dilation of the ER and increased ER stress. C-terminal HTT fragments also bound to and subsequently impaired dynamin 1 activity at ER membranes.
Nuclear protein inclusions (NIs) in HD patients and in a knockin mouse model were shown to be composed primarily of short truncated N-terminal mHTT derivatives, consistent with proteolytic cleavage of mHTT playing a key role in pathogenesis. Since NIs are detectable prior to symptom onset in HD mouse models and in human brains, aggregated mutant proteins forming NIs were thought to be a major cause of cellular dysfunction. However, more recent findings indicate that soluble mutant protein suffices to initiate pathology. Nonetheless, at later stages, NI formation may further aggravate neuronal dysfunction and promote cell death. Support for the latter hypothesis comes from several groups who demonstrated that proteins essential for cellular survival are trapped in NIs. Further, in mouse models, the expression of a truncated mHTT fragment corresponding to exon 1 caused a pathological phenotype more severe than that elicited by full-length mHTT. In addition, cellular models showed that mHTT cleavage product length correlated inversely with its potential to aggregate and cause cell death. Finally, in a neuronal cell model of HD that exhibited time-dependent NI formation from inducible full-length mHTT, NIs were ubiquitinated and contained N-terminal mHTT fragments very similar to the aggregated mHTT breakdown products observed in HD brains, suggesting the activity of proteolytic and other processing steps like those in HD brains.
HTT interacts with both kinesin-1 and dynein motors, as well as over four hundred other proteins, many of which are active in microtubule-based or actin-based intracellular transport. HAP1 (huntingtin-associated protein 1), one of the first HTT interacting proteins to be identified, linked HTT to axonal transport through interaction with the dynein/dynein and kinesin-1 complexes. HTT also bound directly with the dynein intermediate chain. Through these and other interactions, HTT facilitated vesicle transport by decreasing pausing time along microtubules and determining movement direction. Cargo transported by HTT comprised autophagosomes, endosomes, lysosomes, precursors of synaptic vesicles, RNA, protein complexes, Rab proteins, VAMP7, GABA receptors, APP and BDNF (brain-derived neurotrophic factor). In cells or in fly larvae, overexpression of full-length HTT or its N-terminal fragments increased transport efficiency while HTT silencing reduced it. In HD neurons, flies, and mice, mHTT impeded fast axonal transport and interfered directly with trafficking of autophagosomes and BDNF. mHTT bound with higher affinity than HTT to HAP1 and molecular motors, inducing premature detachment of cargo from microtubules and diminishing overall transport efficacy. In human embryonic neural stem cells, selective silencing of the mutant allele restored axonal transport. Striatal neurons depend heavily on trophic support from corticostriatal afferents and loss of BDNF from corticostriatal projecting neurons sufficed to induce neurodegeneration. Conversely, restoring BDNF secretion had a neuroprotective effect on the striatum and corticostriatal synapses. Thus, of all the specific transport cargos associated with HTT, BDNF is likely to be the most essential for preventing corticostriatal neurodegeneration. In summary, both the loss of function of wild-type HTT and the interference in transport caused by mHTT reduced BDNF trophic support to the striatum, making trafficking defects and BDNF insufficiency important elements of HD pathogenesis.
Transcriptional dysregulation has been observed repeatedly in postmortem HD brains and in diverse samples from transgenic HD mouse models. HTT is largely cytoplasmic, but it also occurs in the nucleus. PolyQ tracts like those in HTT are found in transcription factors where they act as transcriptional regulatory domains, mediating binding with transcriptional regulators. HTT binds numerous transcription factors, including CREBBP, NeuroD, SP1, NF-kB and p53. HTT also interacts with transcriptional activators and repressors, including CA150, TAFII130, CtBP, NCOR and NRSF. HTT also binds nuclear receptors, including LXRa, PPARg, VDR and TRa1. Thus, HTT binding of transcription factors and regulators may modulate their nuclear transport. Given the role of HAP1 in intracellular trafficking, it is plausible that the HTT-HAP1 complex participates in nuclear translocation of transcription factors and regulators. Supporting this possibility, HTT bound the NeuroD transcription factor via HAP1, and thereby facilitated its phosphorylation-dependent activation by the mixed-lineage kinase 2 (MLK2). Similarly, HTT influenced the transcription of genes containing the NRSE, a conserved 21-23-bp DNA repressor element recognized by the transcriptional suppressor NRSF. NRSF-regulated genes, such as BDNF, are essential for neuronal development and maintenance. Trafficking of NRSF to the nucleus involved formation of a complex with HTT, HAP1, p150Glued and the NRSF-interacting LIM domain protein (RILP). Thus, activation of BDNF transcription may depend on cytoplasmic sequestration of NRSF by HTT. Finally, HTT regulates chromatin remodeling. For example, both in vitro and in vivo HTT interacts with EZH2 and SUZ12, two components of the polycomb repressive complex 2 (PRC2), thereby promoting its histone H3K27 methylation activity. Through these interactions, HTT can promote and repress gene transcription.
As neurons are terminally differentiated, efficient waste clearance is essential to prevent toxic buildup of protein aggregates. Indeed, HD brains exhibit evidence of defective autophagy. In various HD models, mHTT abnormally activates the autophagy pathway through inactivation of mTOR. However, although the formation of autophagosomes is abnormally high in HD, autophagosome loading is defective, leading to a reduced capacity of cells to degrade aggregated proteins and organelles. HTT may directly regulate selective autophagy via several complementary mechanisms. First, through its scaffolding role in forming the dynein/dynactin/HAP1 complex, HTT regulates the retrograde transport of autophagosomes along axons. Second, HTT silencing reduces localization of optineurin at the Golgi apparatus. Thus, HTT may also regulate the dynamics of autophagosomes/lysosomes through interaction with optineurin/Rab8. Third, striking homology exists between domains of HTT and yeast autophagy proteins Atg23, Vac8 and Atg11. These HTT domains are arranged colinearly from the N- to the C-terminus. In particular, the C-terminal Atg11-like part of HTT interacts with mammalian homologs of the Atg1/ULK1 kinase complex, whereas the Vac8-like central region interacts with Beclin-1. The presence of 11 LC3-interacting repeats (LIRs) within HTT provides further support for a role of HTT in selective autophagy. Such LIR motifs are found in Atg8 family-interacting proteins, including p62 and optineurin, and are key to the capacity of autophagy receptor proteins to link cargos to LC3 and/or GABARAP. The identification of a p62-interacting domain in HTT provided clues to the mechanisms by which HTT regulates both cargo recognition and autophagy induction. HTT binding to p62 facilitates its recognition of lysine 63 ubiquitinylated proteins and thereby promotes cargo loading into autophagosomes. In addition, HTT binding to ULK1 releases it from its interaction with mTOR, leading to the induction of autophagy. This autophagy regulatory function of HTT is conserved in flies and may be fine-tuned through changes in the 3D conformation of HTT. Indeed, the Atg11-like C-terminal domain of HTT interacts with its Atg23-like N-terminal domain, recapitulating the Atg11-Atg23 interaction observed in yeast.
Reactive oxygen species (ROS) are cellular metabolism byproducts, normally cleared by endogenous cellular antioxidant systems. Oxidative stress occurs when oxidants overwhelm antioxidants and ROS damage occurs to proteins, nucleic acids, lipids and other biomolecules. Biochemical and immunohistochemical analysis of postmortem HD brains and HD mouse brains revealed elevated markers of oxidative stress — protein carbonylation, lipid peroxidation and DNA damage. Consistent with this, intracellular mHTT aggregation directly elicited ROS production in a mechanism dependent on glutamine repeat length. Additionally, mHTT induced ROS production in mitochondria by direct interaction with mitochondrial proteins. For example, inhibition of mitochondrial protein import by binding of mHTT fragments to the TIM23 (translocase of the inner membrane 23) complex altered the mitochondrial proteome, thereby inducing ROS production. Finally, mHTT interacted with gp91, the major membrane NADPH-oxidase-2 subunit and a major ROS source in HD. Induction of oxidative stress by mHTT occurred not only via promotion of ROS production but also by impairment of antioxidant systems. Thus, cells expressing mHTT exhibited decreased glutamate-cysteine ligase and glutathione synthetase activities, suggesting decreased de novo synthesis of glutathione. Further, mHTT inhibited MRP1 (multidrug resistance protein 1), a protein responsible for mediating cellular export of glutathione conjugates. In addition, mHTT bound to and disrupted the function of several transcriptional regulators of ROS. For example, mHTT prevented formation of the CREB/TAF4 complex that regulates transcription of PGC-1α, a principal metabolic co-regulator and an important oxidative stress suppressor. Also, mHTT decreased expression of NF-κB, responsible for mediating antioxidant and antiapoptotic signaling in response to ER stress.
(Adapted from: Bates, G. (2005). The molecular genetics of Huntington disease — a history Nature Reviews Genetics 6(10), 766-773; Saudou, F., Humbert, S. (2016). The Biology of Huntingtin Neuron 89(5), 910-926; Lunkes, A., Lindenberg, K., Ben-Haı̈em, L., Weber, C., Devys, D., Landwehrmeyer, G., Mandel, J., Trottier, Y. (2002). Proteases Acting on Mutant Huntingtin Generate Cleaved Products that Differentially Build Up Cytoplasmic and Nuclear Inclusions Molecular Cell 10(2), 259-269; El‐Daher, M., Hangen, E., Bruyère, J., Poizat, G., Al‐Ramahi, I., Pardo, R., Bourg, N., Souquere, S., Mayet, C., Pierron, G., Lévêque‐Fort, S., Botas, J., Humbert, S., Saudou, F. (2015). Huntingtin proteolysis releases non‐polyQ fragments that cause toxicity through dynamin 1 dysregulation The EMBO Journal 34(17), 2255-2271; Abramov, A., Potapova, E., Dremin, V., Dunaev, A. (2020). Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration Life 10(7), 101; Vitet, H., Brandt, V., Saudou, F. (2020). Traffic signaling: new functions of huntingtin and axonal transport in neurological disease Current Opinion in Neurobiology 63(), 122-130; and Srinageshwar, B., Petersen, R., Dunbar, G., Rossignol, J. (2020). Prion‐like mechanisms in neurodegenerative disease: Implications for Huntington's disease therapy STEM CELLS Translational Medicine 9(5), 559-566)