Tau
Background
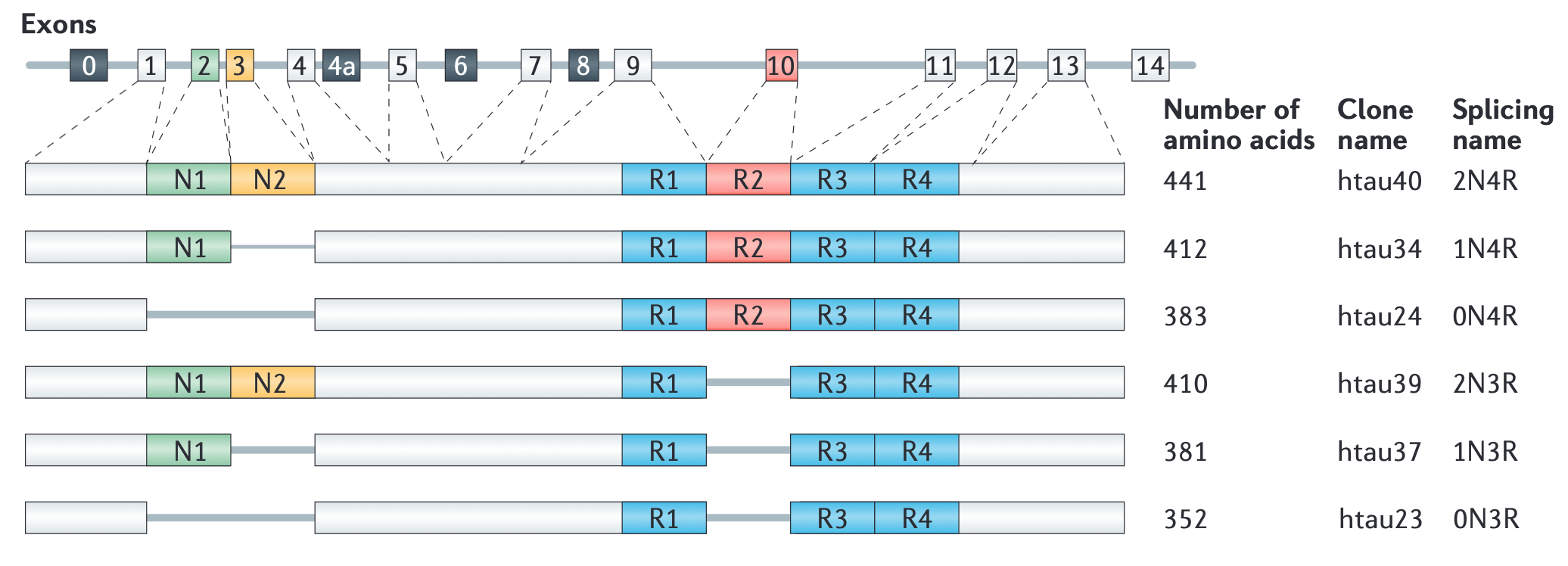
The MAPT gene on chromosome 17q21 encodes tau, a 440 amino acid protein belonging to the microtubule-binding protein family. tau has long been known for its physiological role in regulating microtubule assembly and stability. The assembly of tau into insoluble, filamentous, and hyperphosphorylated intracellular inclusions is the hallmark of the diseases known as the Tauopathies that include PSP, corticobasal syndrome, most FTDs, CTE, and importantly, AD.
Of tau’s six splicing isoforms, those that have four instead of three microtubule-binding domains (R domains) are more aggregation prone. Interestingly, distinct tau isoforms accumulate in different diseases. Thus, all 6 isoforms are found in AD, 3R isoforms in FTD, and 4R isoforms in PSP and corticobasal degeneration (CBD).

Tau protein is structurally divided into acidic, proline-rich, microtubule-binding repeat and C-terminal regions. The four regions constitute two functional domains: N-terminal projection domain (N-domain) and microtubule-binding domain. Silencing of tau expression in neuronal culture did not trigger neurodegeneration nor prevent axon growth, most likely due to functional compensation by other MAPs. Hence it has been suggested that neurodegeneration in Tauopathies is more strongly correlated with tau gaining toxic functions rather than losing normal ones.
 From: Wang, Y., Mandelkow, E. (2016). Tau in physiology and pathology Nature Reviews Neuroscience 17(1), 22-35.
From: Wang, Y., Mandelkow, E. (2016). Tau in physiology and pathology Nature Reviews Neuroscience 17(1), 22-35.
Hyperphosphorylation is a major contributor to tau aggregation. As the longest tau isoform has more than 80 potential phosphorylation sites, the phosphorylation patterns of tau are highly complex and remain incompletely understood. Under physiological conditions, phosphorylation homeostasis is maintained by transient interactions with kinases and phosphatases. Abnormal tau phosphorylation occurs when the phosphorylation/dephosphorylation cycle is perturbed, resulting in accumulation of hyperphosphorylated, non-microtubule-associated tau proteins. Hyperphosphorylated tau can oligomerize and eventually form fibrils known as paired helical filaments (PHF), which in turn can further polymerize into neurofibrillary tangles (NFTs), the neuropathological hallmark of Tauopathies. However, reversible hyper-phosphorylation of tau without development of pathology has been observed in the brains of hibernating animals indicating that hyperphosphorylation alone may be insufficient to trigger pathological tau aggregation.
As phosphorylation impacts both tau’s physiological microtubule-regulating functions and its pathological aggregation propensity, altered interactions with kinases and phosphatases likely play an important role in the development of Tauopathies. AKT1 was suggested to serve a major role in tau biology as it regulates several tau kinases (such as GSK3-beta and PAR1/MARK2) as well as the protein quality control and degradation machinery through the tau ubiquitin ligase CHIP. Notably, Fyn kinase hyperphosphorylated tau in an Aβ-dependent manner. Given that many of the serine/threonine residues phosphorylated in tau were proline-directed, it is not surprising that interaction with the prolyl cis/trans isomerase PIN1 is an important regulator of tau dephosphorylation. Thus, Pin1 expression was inversely correlated with NFT formation in AD brains and Pin1 knockout mice showed age-dependent neuropathy with hyperphosphorylated tau filaments. JIP1 (c-Jun N-terminal kinase-interacting protein 1) is associated with the kinesin motor protein complex. tau hyperphosphorylation promoted interaction with JIP1 that led to impaired kinesin complex function, thus offering a mechanistic explanation for the frequently observed axonal transport defects in AD.
Another PTM closely linked with tau aggregation is cleavage. There are multiple potential protease cleavage sites along full-length tau. C-terminal truncations of tau that produced N-terminal fragments containing the microtubule-binding region were highly prone to aggregation. Inoculation of mice with C-terminally truncated tau induced aggregation of endogenous tau and development of neurofibrillary pathology.
Yet, another driver of altered tau interactions and aggregation is abnormal subcellular protein localization. Hyperphosphorylation resulted in mis-sorting of axonal tau to the somatodendritic compartment where it interacted directly with the PSD-95-NMDA receptor complex. Further, tau-dependent dendritic sorting of Fyn kinase promoted Aβ-induced, NMDA receptor-mediated excitotoxicity. Finally, mislocalized tau could recruit TTLL6 (tyrosine ligase-like enzyme 6) into dendrites where it triggered spastin to sever microtubules, resulting in dendritic spine degeneration.
Several disease-associated tau mutations altered the tau interactome. FTDP-17 (FTD and parkinsonism linked to chromosome 17)-related tau mutations such as P301L/S, G389R, R406W, G272V, K280 or V337M not only strongly promoted tau-Fyn interaction but also drastically reduced tau-PP2A interaction, resulting in tau hyperphosphorylation and aggregation. Some FTDP-17 tau mutations (for example delta-K280 and P301L) also attenuated microtubule binding of tau, thereby increasing cytosolic tau concentration and indirectly promoting aggregation.
Some tau mutations specifically impaired mechanisms that restore proteostasis. For example, TauP301L disrupted interaction with chaperones and proteasomal proteins. Similarly, C-terminal truncations that remove the tau domain responsible for binding chaperones and proteasomal proteins reduce interactions with the protein degradation machinery and thereby promote cellular accumulation. Highlighting the complexity of functional alterations of the tau interactome in various Tauopathies, Pin1 had opposite effects on TauWT and TauP301L. Under physiological conditions, tau also interacted with lipid membranes and this interaction could be affected by disease-associated mutations as was the case with FTDP-17 mutation R406W which disrupted the interaction between tau and Annexin A2, a membrane cytoskeleton linker.
A central tenet of the modern amyloid cascade theory of AD etiology is that Aβ acts upstream of tau aggregation but tau is responsible for most neurotoxic effects and behavioral deficits associated with disease progression, at least in mouse models. The importance of tau for the cytotoxic, dystrophic, and functional effects of Aβ is supported by many in vitro and in vivo experiments as well as by computational modeling. The presence of Aβ pathology in the human brain also enhanced the seeding potential of tau. When tested in an in vitro seeding assay, tissue homogenate from human brains with “tauopathy but no plaques” was less efficacious at seeding tau aggregation than was homogenate from brains with “tauopathy and plaques”. Moreover, the presence of tau pathology exacerbated Aβ deposition in transgenic mice. Thus, instead of Aβ acting exclusively upstream of tau, Aβ and tau may form a positive reinforcing loop.
(Adapted from: Yan, X., Uronen, R., Huttunen, H. (2018). The interaction of α-synuclein and tau: A molecular conspiracy in neurodegeneration? Seminars in Cell & Developmental Biology 99(), 55-64; Valera, E., Spencer, B., Masliah, E. (2016). Immunotherapeutic Approaches Targeting Amyloid-β, α-Synuclein, and tau for the Treatment of Neurodegenerative Disorders Neurotherapeutics 13(1), 179-189; Abramov, A., Potapova, E., Dremin, V., Dunaev, A. (2020). Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration Life 10(7), 101; McAllister, B., Lacoursiere, S., Sutherland, R., Mohajerani, M. (2020). Intracerebral seeding of amyloid-β and tau pathology in mice: factors underlying prion-like spreading and comparisons with α-synuclein Neuroscience & Biobehavioral Reviews 112(), 1-27; Kametani, F., Hasegawa, M. (2018). Reconsideration of Amyloid Hypothesis and tau Hypothesis in Alzheimer's Disease Frontiers in Neuroscience 12(), 25; and Wang, Y., Mandelkow, E. (2016). tau in physiology and pathology Nature Reviews Neuroscience 17(1), 22-35)
Product List
4R-Tau antibody
Antibodies that specifically recognize 3R tau and 4R tau are now commercially available and used for biochemical and histological analysis. Recently, it has become clear that deamidation of Asn279 of 4R tau affects the reactivity of antibodies.This antibody was produced as an antibody that specifically recognizes the 4R tau isoform regardless of Asn279 deamidation, and was reported to be useful for biochemical and histological analysis.
| Product name | Specificity | Catalog Number |
|---|---|---|
| Anti 4R-Tau pAb (Rabbit, Antiserum) | Human, Rat, Mouse | CAC-TIP-4RT-P01 |
Tau antibodies
α-Synuclein Fibrils are confirmed maintaining seed activity in vitro by the Thioflavin T assay.
| Product name | Specificity | Catalog Number |
|---|---|---|
| Anti Tau 354-369 (TauC4) pAb Antibody | Human, Rat, Mouse | TIP-TAU-P01 |
| Anti Tau 360-380 pAb Antibody | Human, Rat, Mouse | TIP-TAU-P02 |
| Anti Tau-N pAb | Human | TIP-TAU-P03 |
| Anti Tau-C pAb | Human, Rat, Mouse | TIP-TAU-P04 |
| Anti-Tau pS199 pAb | Human, Rat, Mouse | BDX-4BDX-1502S |
| Anti-Tau pS199 (clone 2H9) mAb | Human, Rat, Mouse | BDX-4BDX-1501S |
| Anti Tau, phospho Ser416 pAb | Human, Rat | KAL-KR076 |