Navigation
Neurodegeneration Products Dashboard
Aβ
tau
α-synuclein
TDP-43
C9ORF72(this page)
Huntingtin
Ordering Information
Neurodegeneration-related Flyers and Brochures
Antibodies for Neurodegeneration Research (TDP-43, 4R-tau, C9ORF, Synuclein)
PDF Download
Oxidative Stress Markers
mAbs and Selection Guide
PDF Download
Autophagy/Mitophagy Antibodies
PDF Download
Glutamate and NMDA related
Dopamine related
Cholesterol
Ganglioside
Glucosylceramide
Sphingosine
Mitochondria related
Heat shock
Oxidative stress
DNA damage
Lipid peroxidation
Protein carbonylation
Senescence
Transcription and Chromatin
Autophagy
Nucleocytoplasmic transport
Prion protein
APOETREM2
Progranulin
SOD1
FUS
Humanin
Gfap
Paraoxonase
CD36
Dysbindin
BDNF
VEGF
PARP
PPAR gamma
Ubiquitin
Proteasome
Proteases
Caspases
Peptidyl-prolyl cis-trans isomerases
NADPH oxidases
Dyneins
Kinesins
Kinases and Kinase inhibitors
Signaling molecules
Nuclear Hormone Receptors
Products for Neurodegeneration Research: C9orf72
C9orf72 Antibodies and Related Products(click catalog number for product and ordering information)
| Catalog Number | Product Name | Specificity | Size |
| CAC-TIP-C9-P01 | Anti C9orf72 (Poly-GA) pAb (Rabbit, Antiserum) | Human | 50UL |
| CAC-TIP-C9-P02 | Anti C9orf72 (Poly-GR) pAb (Rabbit, Antiserum) | Human | 50UL |
| CAC-TIP-C9-P03 | Anti C9orf72 (Poly-GP) pAb (Rabbit, Antiserum) | Human | 50UL |
| CSB-EL004345HU | Human C9orf72 ELISA Kit | Human | 1x96 rxns |
| UBI-68-0053-100 | TBK1 (human; full length), pAb | Human | 100UG |
| UBI-68-0054-100 | TBK1 pSer172 (human; residues 168 – 177), pAb | Human | 100UG |
| UBI-66-0016-050 | TBK1 [GST-tagged] | Human | 50UG |
In 2011, an intronic GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in the C9ORF72 gene was discovered to be the most frequent cause of familial ALS (40%) and FTD (25%); it also accounted for some sporadic cases (ALS: 8%; FTD: 5%). C9FTD/ALS is the collective term for C9ORF72-associated diseases with clinical features of FTD, ALS or both. Patients with C9ORF72 mutations who have a predominant ALS syndrome can still exhibit cognitive/behavioral features consistent with FTD and, conversely, those with a predominant FTD syndrome can show pathology in the motor system. Indeed, ALS and FTD can occur in the same family and many patients develop signs of both diseases. C9ORF72 HREs have also been identified as a rare cause of other neurodegenerative diseases, including AD, PD, PSP, ataxia, corticobasal syndrome, Huntington disease-like syndrome and Creutzfeldt–Jakob disease. Please scroll to the ORDERING section to explore CosmoBio USA's C9ORF72 reagents that have proven helpful in the study of neurodegenerative diseases.
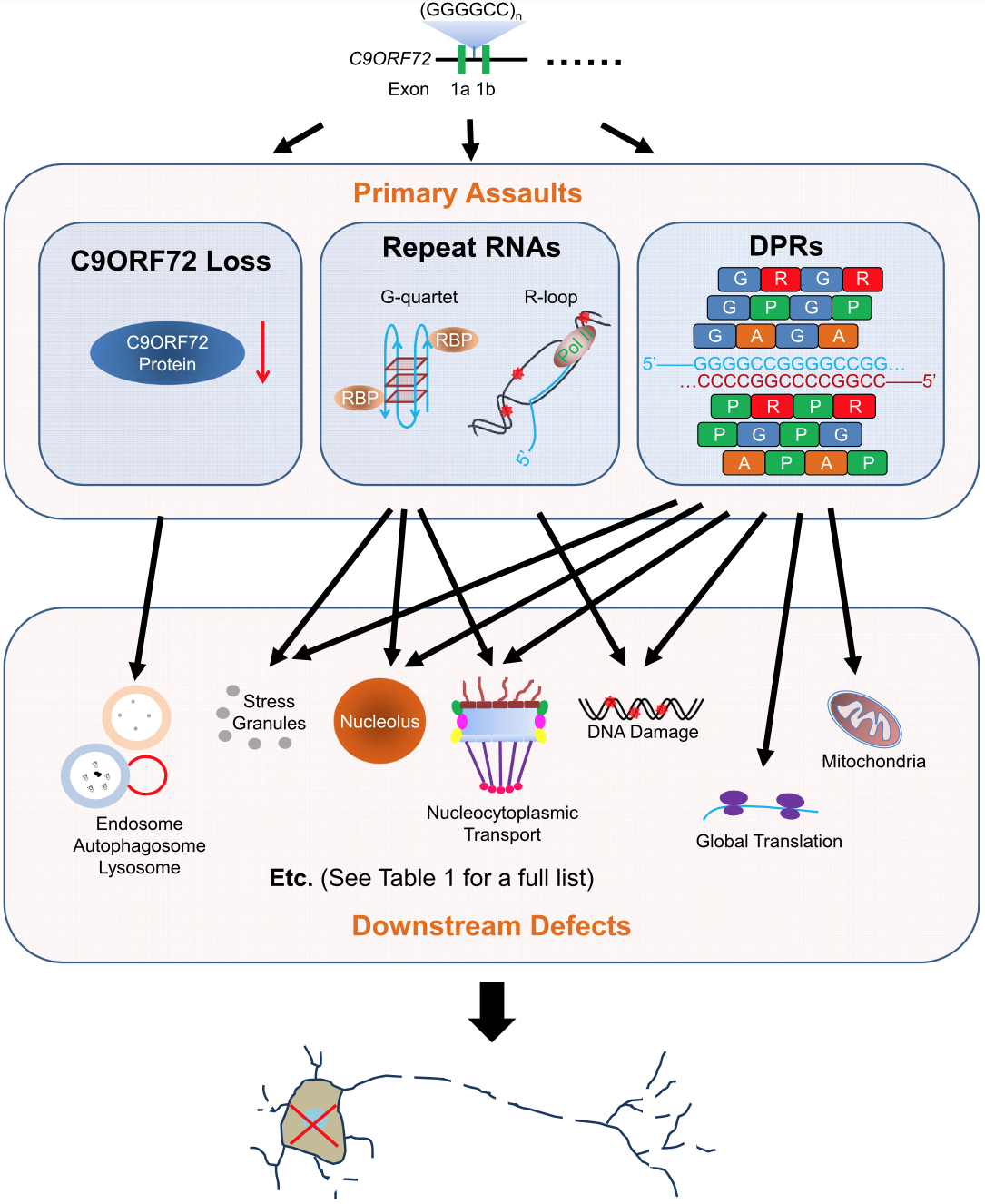 From: Tang, X., Toro, A., T.G., S., Gao, J., Chalk, J., Oskarsson, B., Zhang, K. (2020). Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD Molecular Neurodegeneration 15(1), 34.
From: Tang, X., Toro, A., T.G., S., Gao, J., Chalk, J., Oskarsson, B., Zhang, K. (2020). Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD Molecular Neurodegeneration 15(1), 34.
C9ORF72 is structurally related to members of the RabGEF (rab guanine nucleotide exchange factor) family and functioned as a RabGEF when complexed with two other proteins, WDR41 and SMCR8. RabGEFs activate Rab proteins, a group of small GTPases that regulate membrane trafficking in cells. The C9ORF72-SMCR8 complex promoted the exchange of GDP to GTP, converting inactive GDP-bound RAB8A and RAB39B into active GTP-bound forms, thereby promoting autophagosome maturation. Consistent with these findings, C9ORF72 colocalized to autophagosomes, endosomes and lysosomes. C9ORF72 also activated autophagy via ULK1, a target protein of TBK1. Since loss of TBK1 can also cause ALS and FTD, these data suggested that impaired autophagy may be a common pathomechanism in both C9ORF72- and TBK1-mediated ALS/FTD. In agreement with these data, C9ORF72 overexpression activated autophagy, as indicated by upregulated autophagosome formation. Additionally, in cultured neurons, siRNA against C9ORF72 disrupted autophagy and endocytosis, leading to subcellular aggregation of p62 and/or TDP43. C9ORF72 also regulated actin dynamics in motor neurons by inhibiting the GTP-binding activity of ARF6, leading to ARF6 inactivation, thereby reducing LIMK1 and LIMK2 kinase activity and impairing phosphorylation-dependent inactivation of cofilin. Further, C9ORF72 positively regulated axonal extension and growth cone size in spinal motor neurons. Finally, C9ORF72 plays a role within the hematopoietic system in restricting inflammation and in the development of autoimmunity. Despite this evidence for pleiotropic roles of C9ORF72 in autophagy, membrane trafficking, cytoskeletal organization and immune function, knockouts of mouse C9ORF72 (98% homologous to human) failed to recapitulate ALS or FTD disease. Thus, C9FTD/ALS may be caused by an HRE-dependent gain-of-function mechanism rather than C9ORF72 loss-of-function. Nevertheless, given the role of C9ORF72 protein, as described above, in multiple pathways previously implicated in FTD and ALS, C9ORF72 haploinsufficiency is likely to play an important role in C9FTD/ALS pathogenesis.
How the C9ORF72 HRE causes neurodegeneration is not fully understood. The pathological repeat-length threshold has not been clearly defined. However, the vast majority (>95%) of neurologically healthy individuals have 11 or fewer hexanucleotide repeats, while most C9FTD/ALS patients exhibit repeat lengths of at least 30; and some contain larger expansions ranging from hundreds to thousands of repeats. Two competing but non-exclusive gain-of-function pathogenesis mechanisms for HREs are actively being explored: toxicity from C9ORF72 HRE RNAs and toxicity from dipeptide repeat (DPR) proteins produced by non-ATG (RAN) translation of C9ORF72 HRE RNAs. As RAN translation occurs bidirectionally in all three reading frames, five different DPR species can be produced from G4C2 RNA repeats: poly(GA) and poly(GR) from the sense transcript, poly(PA) and poly(PR) from the antisense transcript, and poly(GP) from both the sense and antisense transcripts.
C9ORF72 repeat RNA foci are widely distributed across the CNS of C9FTD/ALS patients, predominantly within cellular nuclei in the frontal and motor cortices, hippocampus, cerebellum, spinal cord, motor neurons, occasionally in interneurons and sporadically in the cytoplasm. The frequency of frontal cortex neurons expressing sense and antisense C9ORF72 RNA foci was, respectively, ~37% and ~26%; with ~14% expressing both. Less frequently, RNA foci were detected in glia — astrocytes, microglia, astrocytes and oligodendrocytes. G4C2 repeat RNA formed secondary structures in vitro that included hairpins and highly stable G-quadruplexes. These and other secondary structures (DNA-RNA heteroduplexes, RNA duplexes and i-motifs) arising from sense and antisense repeat RNA and DNA sequences are likely to sequester key RNA-binding proteins (RBPs). Pleiotropic effects of such RBP sequestration may underlie pathological defects observed in C9FTD/ALS. For example, in developing zebrafish, expression of both sense and antisense C9ORF72 RNA repeats caused a motor axonopathy phenotype; DPRs were not detected in this model. Primary cortical and motor neurons transfected with a GFP construct interrupted by an artificial intron containing an expanded G4C2 repeat exhibited nuclear RNA foci and reduced survival; dot blots and immunocytochemistry failed to reveal DPRs. In a D. melanogaster model, expression of a construct comprising 30 G4C2 repeat units, each separated from the next by a 6 bp (CTCGAG) sequence, caused degeneration of eye and motor neurons; again, DPRs were not detected. Together these results suggest that repeat RNAs possess pathogenic activity. An important caveat to these studies is the possibility that insufficient sensitivity of detection methods prevented appreciation of a role for specific DPR species; indeed, poly(GR), for example, can be difficult to detect.
DPR inclusions are typically p62-positive, TDP43-negative and can consist of more than one of the 5 individual peptide species. Neuronal DPRs most commonly form cytoplasmic inclusions but can also exist as neuritic inclusions or as ‘pre-inclusions’ that appear as diffuse cytoplasmic staining. Occasionally, inclusions have been observed in the nucleus where they are sometimes associated with the nucleolus. Studies in neuronal cell lines, primary neuronal cultures and non-neuronal cell lines indicated that arginine-rich peptides — poly(PR) and poly(GR) — were the most cytotoxic of the five DPRs. Synthetic poly(GR) and poly(GA) were toxic to primary neurons. In addition, cryo-electron tomography revealed that poly(GA) formed twisted ribbon structures that sequestered the 26S proteasome, suggesting a role in proteostasis. Synthetic poly(GR) and poly(PR) were highly toxic when exogenously applied to cultured human astrocytes. Poly(PR) has a longer half-life than poly(GR) that may explain its relative potency in some contexts. In iPSC-derived neurons, poly(PR) overexpression was toxic. And in a zebrafish model, of the 5 DPRs, poly(GR) had the greatest detrimental effect on development, locomotor activity and lifespan. Together, these studies indicated that DPRs can be cytotoxic and established a relative toxicity ranking: poly(PR) > poly(GR) > poly(GA) >> poly(PA)/poly(GP).
Two D. melanogaster studies implicated DPRs rather than repeat RNAs as the disease-causing neurotoxic entity. In the first study, HRE overexpression in eyes or adult neurons led to neurodegeneration but this effect was inhibited when repeats were interrupted in each reading frame by stop codons that prevented DPR translation. The second study involved ubiquitous overexpression of a construct comprising human C9ORF72 sequence interrupted by an intron containing 160 G4C2 repeats. In neurons and glia, this intron was spliced out and transcribed to produce many G4C2 repeat transcripts that accumulated in sense RNA foci. Importantly, in this version of the model, DPRs were undetectable and there was no evidence of either neurodegeneration, lifespan reduction or widespread transcriptomic changes. However, elevation of transgene expression in this model caused DPR level to become detectable and lifespan to decrease, while sense RNA focus frequency remained unchanged, suggesting that neurodegeneration required DPRs rather than RNA foci. It should be noted that, although the stop codon-interrupted repeat RNA sequence in the first study formed the same G-quadruplex secondary structure as uninterrupted C9ORF72 repeat RNA, the tertiary and quaternary structures formed were not necessarily identical and may have affected the dynamics and specificity of RBP sequestration. This caveat does not apply in the second study where no toxicity was observed despite evidence of multiple RNA foci from stop codon-free C9ORF72 repeat sequences. However, it cannot be ruled out that the functional potency of each repeat RNA focus may have increased with elevated repeat RNA transcription. Taken together, these studies suggest that DPR can cause cellular toxicity and neurodegeneration. Nevertheless, it is likely DPRs and repeat RNAs interact in complex ways in the pathogenesis of C9FTD/ALS.
C9ORF72 mutations can lead to TDP43 inclusions in both ALS and FTD, implying a final common pathway in these diseases. Indeed, most ALS cases and approximately 50% of FTD cases are characterized by neuronal and glial inclusions consisting of the RNA-binding protein TDP43. TDP43 pathology is evident in various brain regions, including the frontal, temporal and primary motor cortices, hippocampus, basal ganglia, amygdala, thalamus and midbrain. TDP43 pathology is likely to be downstream of DPR pathology, probably explaining why it correlates more closely with neurodegeneration. This idea is consistent with downstream effects on TDP43 in some experimental models expressing expanded repeats or pure DPRs, suggesting that an HRE-related gain-of-function mechanism is linked to TDP43 aggregation. For example, adeno-associated virus-mediated CNS overexpression of a cluster of 66 G4C2 repeats led by 6 months to neuronal loss, motor and behavioral phenotypes, RNA foci, DPRs and phospho-TDP43 inclusions. Using the same adeno-associated viral approach, mice overexpressing CNS poly(GA) developed similar neurodegeneration with motor, cognitive and behavioral phenotypes, associated with neuronal cytoplasmic inclusions of fibrillar poly(GA), but no phospho-TDP43 inclusions. As the poly(GA) model failed to fully recapitulate the TDP43 phenotype of the G4C2 repeat mouse, phospho-TDP43 pathology likely requires DPR species other than poly(GA).
Indeed, poly(PR) and poly(GR) have been shown to interact with TDP43, an RBP that, like many others, contains a low complexity domain (LCD). LCD-containing proteins can undergo a process of liquid-liquid phase separation (LLPS) whereby they become compartmentalized in membraneless organelles such as nucleoli and stress granules (SGs). These organelles possess various functions, including facilitation of RNA and RBP assembly into ribonucleoproteins important for RNA metabolism. Mutations in the LCD domains of several RBPs involved in ALS (including FUS and TDP43) can alter LLPS dynamics, leading to aggregation and fibrillization. Primary cortical neurons that overexpressed poly(PR) formed nuclear poly(PR) aggregates and exhibited fewer cytoplasmic P-bodies and more SGs. A 2018 study of patient-derived neurons and in vivo models showed that poly(PR) and poly(GR) induced assembly of SGs that sequestered nucleocytoplasmic transport (NCT) factors and thereby impaired NCT. Suggesting the possibility of a positive feedback loop, poly(PR) and poly(GR) also impaired splicing of RanGAP, an essential NCT regulator. Other interactome studies using nuclear extracts suggested that poly(PR) and poly(GR) associated with the spliceosome component U2 snRNP, blocked spliceosome assembly and disrupted splicing. Together these findings implicate altered LLPS in C9FTD/ALS pathogenesis.
In addition to effects on NCT and splicing, the C9ORF72 HRE is known to perturb translation, mitochondrial function and immune function. In cell lines, overexpression of poly(PR) and poly(GR), but not poly(GA), inhibited protein translation by binding to mRNA, impeding its access to the translation machinery. In a second cell line study, (G4C2)31 expression caused translational inhibition, accompanied by increased SG formation and nuclear accumulation of poly-A mRNA and PABPc, a protein that facilitates cytoplasmic mRNA translation. It is possible that LLPS mediated by G4C2 repeat RNA or DPRs may have caused nuclear PABPc sequestration. When applied exogenously to astrocyte cultures, poly(GR) and poly(PR) 20mers accumulated in the nucleolus and led to aberrant splicing and impaired ribosomal RNA maturation. In cultured cells, primary neurons, iPSC-derived neurons and D. melanogaster, poly(PR) and poly(GR) colocalized with nucleoli exhibiting abnormal morphology. Importantly, the brains of individuals with C9ORF72-associated FTLD exhibited striking nucleolar volume changes, with smaller neuronal nucleoli overall but enlarged nucleoli in neurons containing poly(GR) inclusions. Mitochondrial morphology is disrupted in C9ALS/FTD fibroblasts. Consistent with these findings, in iPSN and/or mouse models, poly(GR) has been shown to disrupt mitochondrial function by sequestering key mitochondrial proteins. Moreover, poly(GR) and poly(PR) have also been shown to disrupt axonal transport of mitochondria. Finally, C9ORF72 HREs in innate immune cells result in loss-of-function toxicity via impairment of cellular homeostatic processes including autophagy. C9ORF72 expression is particularly high in microglia, resident innate immune cells of the brain. C9ORF72 mutations may have consequential effects on the regulation of synapses by microglia and may cause persistent microglial activation that has a pathogenic effect, exacerbating the progression and development of C9FTD/ALS. Excessive or chronic activation of the innate immune system has been linked to increased cellular stress and degeneration of motor neurons.
(Adapted from: Tang, X., Toro, A., T.G., S., Gao, J., Chalk, J., Oskarsson, B., Zhang, K. (2020). Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD Molecular Neurodegeneration 15(1), 34; and Hutten, S., Dormann, D. (2019). Nucleocytoplasmic transport defects in neurodegeneration — Cause or consequence? Seminars in Cell & Developmental Biology 99(), 151-162; Cook, C., Wu, Y., Odeh, H., Gendron, T., Jansen-West, K., Rosso, G., Yue, M., Jiang, P., Gomes, E., Tong, J., Daughrity, L., Avendano, N., Castanedes-Casey, M., Shao, W., Oskarsson, B., Tomassy, G., McCampbell, A., Rigo, F., Dickson, D., Shorter, J., Zhang, Y., Petrucelli, L. (2020). C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy Science Translational Medicine 12(559); and Balendra, R., Isaacs, A. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease Nature Reviews Neurology 14(9), 544-558)